Nanomedicine, Volume IIA: Biocompatibility
© 2003 Robert A. Freitas Jr. All Rights Reserved.
Robert A. Freitas Jr., Nanomedicine, Volume IIA: Biocompatibility, Landes Bioscience, Georgetown, TX, 2003
15.2.3.3 Immunoglobulins (Antibodies)
The “humoral” (B-lymphocyte) branch of the specific immune system (Section 15.2.3.1.2) responds to the presence of different antigens (such as foreign molecules) in the body by manufacturing large numbers of complementary antibodies capable of binding to those antigens, then releasing these antibodies into the bloodstream after a delay of up to 4-10 days. An antibody molecule has two* principal functions: (1) to recognize and bind to an antigen, and (2) to assist in the destruction and elimination of that antigen [353]. This dichotomy of function is reflected in structure because every antibody molecule has discrete domains that participate in one of these two functions – a variable region to enable recognition, and an effector (“common”) region to enable elimination. Specific antibodies can be made against virtually any foreign chemical group (but see discussion of immunogens in Section 15.3.7).
* In late 2002, a possible third function was reported: evidence that antibodies can directly catalyze the production of highly active forms of oxygen (likely including ozone) that may not only kill bacterial pathogens directly but might also promote inflammatory and other immune responses [6018].
Antibodies are a class of glycoproteins called immunoglobulins, abbreviated Ig, which collectively comprise ~25% of total noncellular blood plasma protein. Each monomeric antibody molecule has an approximate molecular weight of 150-190 kD [1753], and there are ~1020 antibody molecules in every adult human body [1767]. In general, antibodies can be found: (1) inside cytoplasmic membrane-bound compartments such as ER (endoplasmic reticulum) and Golgi, (2) on the surface of B-cells, (3) in blood plasma, (4) in the interstitial fluid of tissues where secreted antibodies from B-cells accumulate, (5) in secretory fluids such as mucus and breast milk into which certain types of antibody molecules are specifically transported, and (6) on the surface of certain immune effector cells such as mononuclear phagocytes, NK cells, and mast cells, which do not synthesize antibody but have specific receptors for binding antibody molecules [5491].
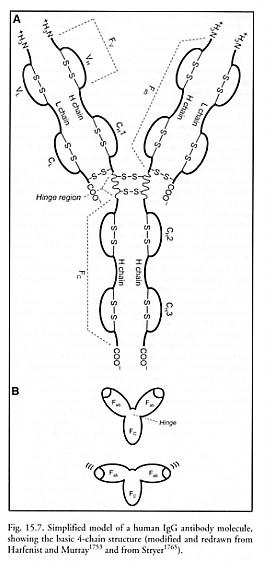
The most common human antibody molecules consist of two identical ~50-70 kD “heavy (H) chains” that are noncovalently joined in the lower half (the “constant,” “effector” or “Fc” region) and separated in the upper half (the “antigen-binding” or “Fab” region), forming a “Y” shape (Figure 15.7). Each heavy chain is ~446 amino acids long [1765], and has a hinge region of 10-60 amino acids near the middle, allowing the two upper arms of the Y to swing and rotate relatively freely. Two identical ~23 kD “light (L) chains” are attached to the two heavy chain upper arms, one per arm, with each L chain ~214 amino acids long (Figure 15.7). The L chains and H chains are synthesized as separate molecules, then assembled into mature Ig molecules inside the B cell or plasma cell, at production rates up to 1000-3000 Ig molecules/sec-cell [1754, 1755].
The top halves of each upper arm on each chain are designated as the variable (VL and VH) domains. These domains are ~117 amino acids in length and have sequences that differ from other antibody molecules, enabling the binding of a specific antigen and allowing molecular recognition to occur. Antibody specificity for antigen varies for different epitopes (active binding regions on antigens) and is somewhat degenerate – that is, a given antibody can react with more than one epitope, provided these epitopes are closely related structurally. A given antigen may contain several unrelated epitopes; however, an antibody cannot have two different antigenic binding sites.
Each clone of antibody-producing cells makes a unique antibody. The variable domains from different humans also contain unique allotypic amino acid sequences. But each variable domain also includes 3 light-chain “hypervariable” regions and 3 heavy-chain “hypervariable” regions [5491] where most of the epitope-specific sequence variation occurs. Each of these widely separated hypervariable regions consists of only 5-10 (or in one case 15) contiguous amino acids, so most of the variability in both L and H chains is restricted to about 15-30 amino acids per chain. Note that the variable domains are not simple linear sequences of amino acids, but rather form globular regions with secondary and tertiary structure in order to effect binding of specific antigens [1753]. The antigen binding sites are pockets formed when the hypervariable regions fold into close proximity, producing a 3D structure with a surface complementary to the 3D surface of the bound antigen [5491]. Antibody configurations are produced by somatic recombination of ~78 light-chain and ~84 heavy-chain gene segments [1758], which, along with other sources of variability, allow for up to ~109 chemically distinct antibody receptor specificities [353, 354], although only ~107 specificities are found in a single individual [1756].
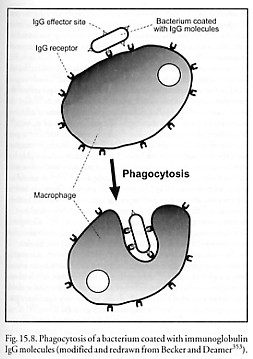
The remainder of each chain is designated as constant (CL and CH) domain of several types, which varies relatively little among immunoglobulin molecules of similar class (isotype) and thus determines the class of antibody. The Fc or effector region of each molecule can be recognized by complementary antibody receptors present on the outer surface of monocytes, neutrophils, eosinophils, NK cells and macrophages (IgG1 and IgG3 only). This permits phagocytic elimination of the antibody-bound antigen on, say, a bacterial outer membrane (Figure 15.8). Because each antibody molecule has two or more antigen binding sites (Figure 15.9), antigens can be crosslinked by antibodies into chains, lattices, and networks, forming immune complexes that facilitate phagocytosis and complement activation.* The formation of immune complexes on the surface of a biochemically active antigen can block its binding site, thus inhibiting its biological activity.
* The reactions of antibody with multivalent insoluble particulate antigens results in the crosslinking of the various antigen particles by the antibodies, eventually producing clumping or agglutination of the antigen particles by the antibodies [1760] – the basis for the standard agglutination test.
There are 5 classes of immunoglobulin molecules [353, 1753, 1756-1760]:
Immunoglobulin G (IgG) is the most abundant antibody in the blood, as well as the most common antibody produced in late primary and in secondary immune responses. IgG is a 150 kD monomer (~76% of total serum Ig, or ~13.5 mg/ml) with a half-life of ~3 weeks. It is also distributed in extracellular fluid and is present in milk, and maternal IgG is the only Ig that normally crosses the placenta. IgG binds to the surface of somatic and microbial cells, which allows those cells to be phagocytosed or killed by cytotoxic cells (Figure 15.8) – along with IgM, IgG is the primary activator of the complement system. IgG also binds complement via an Fc receptor present in the constant region of the heavy chain. S. Flitman notes that only IgG crosses the blood-brain barrier (0.1% of all IgG is in the CNS compartment at all times.) There are four subtypes with differing activities and concentrations: IgG1 (~9 mg/ml), IgG2 (~3 mg/ml), IgG3 (~1 mg/ml), and IgG4 (~0.5 mg/ml).
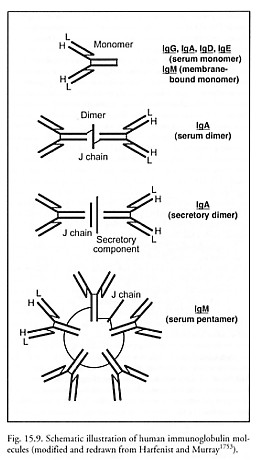
Immunoglobulin A (IgA) is the most important class of antibody found in secretions such as tears, sweat, saliva, colostrum and breast milk, and in mucus secretions of the bronchial, gastrointestinal and urogenital tracts, where it is present as “secretory IgA”, a 335 kD dimer consisting of two Y-shaped units linked together at the foot of each Y by a transverse 15-kD “J chain”. A 70-kD polypeptide called secretory component is attached to the Fc portion after the dimer has been endocytosed into the lumen of secretory tissue, so a ~400 kD complex is normally found in secretions with a half-life of ~6 days. Secretory IgA plays an important role in host defense against viral and bacterial infections by binding to microbes and thus blocking their attachment and transport across mucosa, and provides passive immunity to breast-fed babies. “Serum IgA”, a simple 160 kD Ig monomer, is the second most common Ig in the blood, constituting ~15% of serum Ig (~2.7 mg/ml). There are two subtypes: IgA1 and IgA2.
Immunoglobulin M (IgM) is the first class of antibody to be produced during B cell development. IgM is also the major class of antibody secreted first into the bloodstream during a primary immune response. IgM is found mainly in the intravascular compartment and on B-cell surfaces. It is normally absent from organs and tissues, and usually doesn’t cross the placenta. Carbohydrate antigens such as blood group substances stimulate IgM, and IgM efficiently activates the complement system, facilitating the death of invading microbes. (IgM, IgG1, IgG2, and IgG3 are complement-fixing antibodies; the rest of the Ig’s are non-complement-fixing antibodies.) Membrane-bound IgM is the usual ~175 kD Y-shaped monomer, hydrophobic at one end to remain anchored in cell membrane where it serves as a surface receptor. Serum IgM molecules are soluble (hydrophilic) pentamers bound together by disulfide bridges plus a 15-kD J chain, with total molecular weight of ~900 kD, constituting ~8% of all serum Ig (~1.5 mg/ml) with a half-life of ~5 days.
Immunoglobulin D (IgD) is a monomeric 180 kD antibody very prominent on mature B-cell surfaces where it is co-expressed with IgM. But IgD is secreted by very few B cells, hence constitutes only ~0.2% of all serum Ig (~0.03 mg/ml) with a half-life of ~3 days. IgD functions as an antigen receptor optimized to efficiently recruit B cells into antigen-driven responses [1761], and can substitute for IgM functions [1762]. Expression of membrane IgD appears to correlate with the elimination of B cells having the capacity to generate self-reactive antibodies, so the major biologic significance of IgD may be the silencing of autoreactive B cells during development [1760].
Immunoglobulin E (IgE) is a monomeric 190 kD antibody that constitutes only ~0.003% of all serum Ig (~0.0005 mg/ml) with a half-life of ~2 days. IgE is important in allergic disease because it binds to the surface of mast cells and basophils. The capacity of IgE to trigger inflammatory reactions, specifically with eosinophils, is also beneficial in the clearance of extracellular parasitic infections. In the presence of specific antigen which ligates (cross-links) two adjacent surface-bound IgE molecules, IgE induces the cell to release granules containing vasoactive amines (e.g., histamine and serotonin) and various allergic-response molecules including leukotrienes, prostaglandins, platelet activating factor, proteases and cytokines, resulting in bronchospasm, vasodilation, smooth muscle contraction, and chemoattraction of other inflammatory and immune cells.
No covalent bonds are formed during the interaction between antibody and epitope (the specific antibody binding portion of an antigen or immunogenic macromolecule) [1760]. Binding forces are relatively weak, consisting mainly of van der Waals, electrostatic, and hydrophobic forces (Section 3.5.1), all of which require a very close proximity between the interacting moieties that is often compared to a lock and key. The low binding energies allow antigen-antibody complexes to be readily dissociated by low or high pH, by high salt concentration, or by chaotropic ions such as cyanides that interfere with the hydrogen bonding of water molecules [1760].
Can the human immune system recognize medical nanorobots? The answer may depend largely upon the composition of the nanorobot exterior surfaces. Pure diamond is generally considered nonimmunogenic – e.g., chemical vapor deposition (CVD) diamond coatings for artificial joints are said to have “low immunoreactivity” [535], and as of 2002 there were no reports in the literature of antibodies having been raised to diamond. Even low molecular weight adamantane-based derivatives yield inherently non-antigenic antiviral drugs, though when incorporated into dipeptide gels these drugs can induce the production of high-titer specific antibodies in rabbits [5561]. Other adamantane derivatives such as rimantadine interfere with and suppress the generation of cellular immune responses [5562].
As for nondiamond carbon materials, graphite-based endoprostheses elicit no immunological reactions [820]. Carbon particles in India ink induce a reaction to human serum IgG only if the particles are pretreated with staphylococcal protein A [863]. On the other hand, carbon black can have a significant adjuvant effect on the systemic specific IgE response to allergen (ovalbumin) in mice [867]. Solubilized (derivatized) C60 and C70 fullerenes can induce the production of specific antibodies [724, 725, 2387-2389], usually by interaction with the combining sites of IgG [725]. It is speculated that highly hydrophobic pure fullerenes would be recognized by antibodies with hydrophobic amino acids in their binding sites [725, 2164] or would interact with donor -NH2 [914] and -SH [915] groups. There are several reports of antibodies being raised to single-walled carbon nanotubes [2164, 2385-2387, 4630]. For example, antibodies raised to C60 in mice strongly bind to single-walled nanotubes [2386]. Computer simulations suggest that it may be possible to build antibodies that selectively bind to nanotubes of a specific diameter or chirality [2164]. As for noncarbon materials, pure sapphire appears reasonably nonimmunogenic, although similar hydrophilic surfaces do adsorb immunoglobulin IgG [543]. Adsorption-induced denaturation of immunoglobulin G (IgG) on Teflon doesn’t lead to complete unfolding into an extended polypeptide chain, but leaves a significant part of the IgG molecule (the Fc fragment) in a globular form [1336]. Crystalline silicon [1769], silica ceramic [1770], PTFE membrane [1771] and Teflon [1772] immunoisolation microcapsules appear to be nonimmunogenic during extended periods of implantation. Various biological materials appear immunologically inert, such as hydroxyapatite [1834].
However, concerted experimental searches for antibodies to diamondoid materials have yet to be undertaken, and experimental failures rarely find their way into the literature. Immunologists usually work on the assumption that the available antibody repertoire is diverse enough to ensure the production of antibodies to virtually any potentially antigenic molecule [1768] (but see Section 15.3.6). Izhaky and Pecht [724] suggest that since fullerenes (and other diamondoid materials) are highly ordered and symmetric molecules for which scant experimental data exists, it might be useful to compare the ability of vertebrate immune systems to respond to analogous non-diamondoid water-insoluble highly-ordered antigens.
For example, water-insoluble crystals introduced into experimental animals are found to be treated as antigens [5035], inducing specific antibodies. Kessler et al [1774] raised monoclonal antibodies (MAbs) specific for crystals of 1,4-dinitrobenzene having well-defined molecular-level structures. These antibodies were so specific they would not bind to the same molecule when it was conjugated to a protein carrier. Antibody binding sites typically span a contact area of 6-9 nm2 [1775, 1776], so an antibody can bind to arrays of 5-20 molecules exposed at the crystal surface [724] like an artificial semaphore presentation array (Section 5.3.6). IgG antibodies isolated from the serum of rabbits injected with crystals of monosodium urate monohydrate or magnesium urate octahydrate evidently bear in their binding sites an imprint of the crystal surface structure because they can act as nucleating templates for crystal formation in vitro with extremely low cross-reactivity, despite the similar molecular and structural characteristics of the two crystals [1777]. Antibody binding to monosodium urate crystals has been known for decades [5037-5039], and viruses have been engineered with a specific recognition moiety for ZnS nanocrystals used as quantum dots [5040]. Interestingly, antibodies specific to in vivo water-ice crystals have even been reported [1773].
Like antigens with ordered multiple epitopes, crystals expose chemically and geometrically distinct surfaces. It is conceivable that different antibodies may recognize distinct faces of a crystal (possibly including diamond or sapphire crystal faces exposed at the surfaces of medical nanorobots) in an interaction similar to that of antibodies for repetitive epitopes present on protein surfaces [724, 5036]. For instance, one MAb to 1,4-dinitrobenzene crystals was shown to specifically interact with the molecularly flat, aromatic, and polar (101) face of these crystals, but not with other faces of the same crystal [1778]. MAbs have also been elicited against cholesterol monohydrate crystals [1779, 5034], one of which [1779] was shown to specifically recognize the crystal’s stepped (301) face. Here, the hydrophobic cholesterol hydrocarbon backbone is exposed on one side of the molecular steps while hydroxyl residues and water molecules are exclusively exposed on the other side. In both cases, crystal-specific antibodies were of the IgM idiotype [724]. This accords with the assumption that (unlike most commonly used antigens) crystals cannot be processed by the antigen presenting cells, hence antibodies must be induced through a T cell-independent path [1780]. Semiconductor-binding [2170, 5040] and calcite-binding [5243] proteins are known that can discriminate among the various crystal faces of the given material and can in some cases alter the pattern of crystal growth [5244]. Sulfur-free gold-binding proteins (GBPs) recognize and noncovalently bind preferentially to the Au (111) crystal surface – GBPs use multiple repeats of 14-30 residue sequences to bind to this surface [2391]. Hyaluronan is believed to be a crystal-binding protein for calcium oxalate monohydrate crystals [5245].
Diamondoid surfaces coated with non-self adsorbed protein monolayers (Section 15.2.2) might prove antigenic, as might protein-based presentation semaphores (Section 5.3.6) that become detached via degradative intracellular chemical processes and whose fragments are subsequently presented at the cell surface by MHC molecules (Sections 8.5.2.1 and 15.2.3.1.2). Avoiding such detachment will be an important design objective for many nanorobot missions. Another concern is that antibodies may be raised against binding sites that are positioned on the nanorobot exterior, e.g., sorting rotor pockets (Section 3.4.2), which are similar to traditional bioreceptors, or manipulator end-effectors (Section 9.3.2). These antibodies could then act as agonists [1783] or antagonists [1781, 1785] for such sites, since MAbs specific to biological binding sites are well known [1781-1785]. This risk may be increased if nanorobot binding sites employ non-self biomolecular components, or, conversely, may be decreased if binding sites employ purely diamondoid rigid structures or self-biomolecule receptors (e.g., whose natural antibodies have likely been eliminated by clonal deletion). This is an additional design constraint that must be addressed experimentally.
If antibodies to nanorobot exteriors can exist in the natural human antibody specificity repertoire, then to avoid immune recognition many techniques of immune evasion (Section 15.2.3.6) may be borrowed from biology, possibly including:
(1) Camouflage. Coat the nanorobot with a layer of “self” proteins and carbohydrate moieties resembling fibroblast, platelet, or even RBC [1788] plasma membrane. Normally, antibodies for these surfaces have already been deleted from the systemic repertoire to avoid autoimmunity, so the coated nanorobot will be theoretically nonimmunogenic. Ideally, an artificial surface would be designed that displays the minimum necessary ligand set to ensure nonimmunogenicity. Presentation semaphores (Section 5.3.6) may be used if the required surface ligand concentration is significantly less than monolayer thickness. The existence of nonimmunogenic autologous cells such as NK [2171], TH1, and malignant cells (via HLA-G expression) [2166] that escape immunosurveillance, and bacteria capable of evading the antibody response [1786-1789], suggests that such nonimmunogenic exteriors are possible. Extended rejection-free allograft survival using a combination of T-cell costimulation inhibitor and anti-CD40 MAb has been demonstrated experimentally in primates [2541]. Personalized nanorobots exhibiting self-MHC receptors (Section 8.5.2.1) on their surfaces would possess a very specific type of camouflage. Autoimmune risk due to unwanted detachment of self moieties from the nanorobot surface, and pathogen borrowing of such detached moieties for the purpose of immune evasion, especially in the case of large localized populations of in vivo devices, should be studied further.
(2) Chemical Inhibition. Nanorobots may slowly secrete chemical substances into the perirobotic environment to make it difficult for Ig molecules to adhere to an otherwise immunogenic nanorobot surface. For example, a >0.01% concentration of sodium dodecylsulfate surfactant destroys almost all antigen-antibody binding [1790], but the emission would have to be very localized to avoid lysing other cells in the vicinity prior to denaturing an antibody. The low pH gastric environment produces poor Ig deposition, allowing H. pylori bacteria to evade humoral defenses [1791]. Bacteria such as Pasteurella multocida [1792], Pseudomonas aeruginosa [1793], and Serratia marcescens [1794] secrete extracellular proteases that can cleave Ig molecules. Covalent pegylation of otherwise antigenic proteins can induce specific tolerance [1766, 1833]. There may be some risk of local inflammation with this approach.
(3) Decoys. Release a cloud of soluble nanorobot-epitope antigens in the vicinity of the nanorobot. This will not affect nanorobot operations because the decoy molecules are noncomplementary to nanorobot surfaces. But the decoys will bind any antibodies specific to the nanorobot epitopes, preventing further antibody activity against the nanorobot [1437]. J.R. Baker notes that this would have to be done cautiously to avoid triggering serum sickness or complement activation. Alternatively, decoy fragments may be loosely bound to the nanorobot surface and jettisoned as soon as a binding event is detected (Sections 4.2.1, 4.2.2, and 4.2.3). This could limit mission duration to the exhaustion time of the onboard supply of decoy molecules. A. Kumar notes that decoy releases would have to be controlled very accurately for all the nanorobots in the body because there is a threshold level of antigen that triggers the immune response.
(4) Active Neutralization. Equip the nanorobot with molecular sorting rotors designed with binding sites similar or identical to the nanorobot epitopes that raised the target antibodies. The target antibodies will bind to rotor pockets and be conveyed inside the nanorobot, whereupon the antibody molecules can be chemically altered (a) to eliminate their troublesome paratopes (sites of epitope attachment) or (b) to defunctionalize their effector region. They would then be released back into the body in a harmless neutralized state, care having been taken to avoid random chemical alterations which might trigger autoimmune responses. Less efficiently (especially when the immune system is fully activated), the ingested target antibody could be chemically degraded or cleaved [1792-1797] into safe peptides suitable for free release, or simply warehoused onboard until the end of the mission. M. Sprintz notes that if the nanorobotic mission is short-term (a few days), then antibody production is not an issue for the first exposure, though subsequent exposure could produce a delayed-type hypersensitivity (DTH) response (Section 15.2.6.1); he suggests also the possible active nanorobotic prevention of memory cell formation.
(5) Tolerization. Nanorobots introduced into a newborn may train the neonatal immune system to regard these foreign materials as “native,” thus eliminating nanorobot-active antibodies via natural clonal deletion [1828-1830]. This process is often called “tolerance induction” and in this example assumes a mature nanomedical technology with well-defined nanorobot surface signatures that will not change over time as the neonate matures into an adult. Pregnant women may develop specific immunological tolerance to fetal antigens and foreign transplant tissue [1831], thus might also become tolerized to nanorobotic antigens introduced during pregnancy. Nonpregnant patients could have tolerance artificially induced via engineered antigen-specific T-suppressor cells [371, 5349-5354] or by other means (Section 15.2.3.4). This approach seems feasible if nanorobots use only a few key surface materials – deactivating immune responses could have serious implications, e.g., failure to recognize a pathogenic microbe due to cross-tolerance.
(6) Clonal Deletion. Once the paratopes of antibodies that bind nanorobots are known, immunotoxin molecules can be engineered that display those paratopes.* Upon injection into the patient, these targeted immunotoxins would bind to all T cell receptors that display this paratope, killing the nanorobot-sensitive T cells [1803-1810]. Engineered immunotoxins may also eliminate all B cells capable of manufacturing antibodies having the proscribed nanorobot-binding paratope [1811-1816], at EC50 concentrations as low as 2.5-70 ng/ml (~0.03-0.8 molecules/micron3) [1814]. Such interventions could be made at the local, lymphatic, or systemic levels [2543]. The end result is that the ability of the immune system to recognize nanorobot epitopes would be selectively eliminated, in effect adding “nanorobot surfaces” to the definition of “self” by a process of artificial clonal deletion against T cells [1817-1823] and B cells [1824-1827].
* Raising artificial MAbs against natural antibodies that react to a nanorobot is another approach, though this is more risky because anti-antibodies [1798] are found in several immunopathological or autoimmune diseases. Antibodies specific for determinants within the variable region of an antibody molecule are known as anti-idiotypic antibodies [1799-1801], but these function as surrogate antigens and might actually stimulate additional anti-nanorobot immune response. Anti-anti-idiotypic antibody responses have also been elicited experimentally [1802].
Last updated on 30 April 2004
{kind=link}
{kind=link}
{kind=link}