Nanomedicine, Volume IIA: Biocompatibility
© 2003 Robert A. Freitas Jr. All Rights Reserved.
Robert A. Freitas Jr., Nanomedicine, Volume IIA: Biocompatibility, Landes Bioscience, Georgetown, TX, 2003
15.2.3.2 Complement Activation
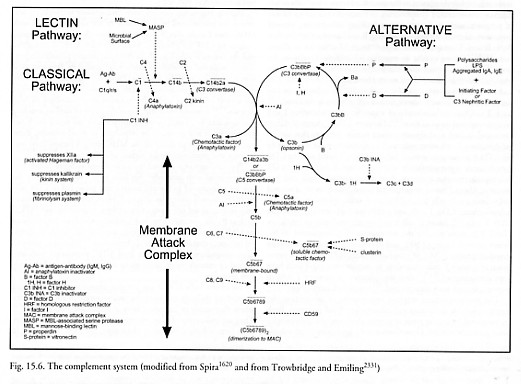
The complement system [1613, 1616-1624, 2331, 2539] consists of a series of ~20 plasma and cell membrane proteins (e.g., named C1-C9, etc.) synthesized mainly by the liver and certain cells of the immune system (e.g., macrophages). These proteins interact in a sequential or regulatory manner, leading (1) to the general promotion of inflammatory reaction, including mediating vascular responses such as histamine release and recruiting phagocytic leukocytes via chemotaxis, and (2) to the lysis of certain kinds of cells and susceptible microorganisms following attachment of the membrane attack complex (MAC) to their plasma membranes (Figure 15.6). The targets of this process may be bacteria, virus-infected human cells, mycoplasmas, spirochetes, protozoa, or tumor cells [1620]. The complement system may follow two different activation pathways* – the classical pathway or the alternative pathway – either of which can initiate the terminal sequence of complement activation which involves assembly of the MAC.
* A third “lectin pathway” for complement activation via foreign carbohydrates on microbial surfaces, leading to cell lysis, has recently been discovered [1608-1613, 5343], with activity similar to that of the classical pathway [1606] but apparently predating it evolutionarily [1607]. As with other pathways, the lectin pathway can be inhibited [5344-5347].
The classical pathway of the complement system is activated by antigen-antibody complexes that have formed on the surface of a target cell. Complement factor C1 (900 kD) binds to the Fc (Section 15.2.3.3) portion of either a single antibody molecule of IgM or to a pair of antibody molecules IgG1, IgG2, or IgG3, in apposition on the surface of the antigen. C1 is a macromolecule composed of C1q (410 kD) and doublets of C1r (85 kD) and C1s (85 kD) linked by Ca++ ions. The C1q component binds to the antibody,* activating C1r and C1s to form C1 which itself has enzymatic activity to cleave C4 (210 kD). The cleavage of C4 releases the C4a (6 kD) fragment into solution and attaches the larger C4b fragment at the site, making the C14b complex, which can now bind C2 (110 kD). Once bound (the process is complete in 5-10 min [1617]), C2 can be cleaved by the C1 complex (or other proteolytic enzymes like trypsin or chymotrypsin). This releases the smaller C2b (35 kD) fragment into solution and leaves the larger C2a (75 kD) fragment attached at the site, making the C14b2a complex. The C14b2a complex is the first of the two forms of C3 convertase.
* A variety of other substances interact directly with C1 and C1q [1626], including negatively-charged polyanionic substances which form a complex with cationic C1q, the most basic serum protein with an isoelectric point of ~pH 9.2. Direct binding of C1q has been shown for polynucleotides, heparin, dextran sulfate, condroitin sulfate, cardiolipin, LPS, the envelopes of some RNA viruses [1617], certain microorganisms and some retroviruses and mycoplasmas [1618]; C1q can be involved in viral lysis initiation [1620].
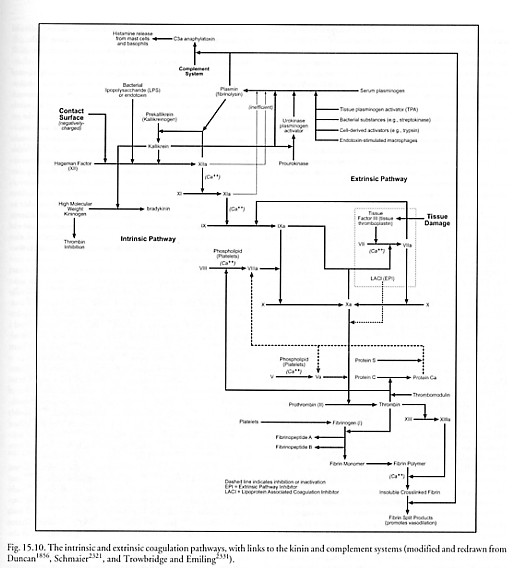
The alternative pathway of the complement system is of greater importance in the initial defense against infection, because it does not depend solely upon the time-consuming production of specific antibody [2331]. It may be activated by contact with the Fab (Section 15.2.3.3) portions of aggregated IgA or IgE, or in some cases the F(ab’)2 portion of IgG antibodies complexed with antigen on the surface of a target cell. More importantly, the alternative pathway may be activated in the absence of antibody complexes by certain foreign molecules such as repeating sugars or proteins, as, for example, plasmin (see Figure 15.10), bacterial lipopolysaccharide (LPS), yeast zymogen, trypanosomes, plant (inulin) polysaccharides, polyanionic substrates (e.g., dextran sulfate), cobra venom factor, heterologous erythrocytes, carbohydrates (e.g., agarose), and many other materials. These substances activate factor P (properdin, 220 kD), activating in turn a C3-convertase amplification loop. In this amplification loop, pre-existing C3b (180 kD) fragments opsonize* (attach to) the target cell. Factor B (93 kD) reversibly binds to a receptor on the surface-bound C3b fragment. Factor D (an enzyme circulating at low concentration in active form, 22 kD [1618, 1675-1677]) cleaves the bound factor B, releasing Ba (30 kD) into solution and leaving attached the larger Bb (63 kD) segment. This makes the C3bBb complex which can cleave many more C3 (195 kD) molecules, some of which bind covalently to the activating surface. However, the C3bBb enzyme dissociates fairly rapidly unless it is stabilized by the binding of activated factor P, creating the C3bBbP complex.** The C3bBbP complex is the second of the two forms of C3 convertase, and completes the amplification loop, resulting in the binding of many more molecules of C3b to the same surface.
* Opsonins are substances that coat foreign antigens, making those antigens more susceptible to recognition by macrophages and other leukocytes and thus increasing phagocytosis of the organism or object displaying those foreign antigens. In effect, opsonins promote cytocarriage (Section 9.4.7) by macrophages. The two main opsonins in human blood are complement and antibodies.
** The C3bBb enzyme can also be stabilized by C3 nephritic factor, an IgG autoantibody directed against an antigen of C3bBb [1620].
The terminal sequence of the complement system actually builds the MAC. This process is triggered when either form of C3 convertase accumulates on the target surface. C3 convertase has specific enzymatic activity to cleave C3, releasing C3a (9 kD) into solution and attaching the larger C3b fragment to the C3 convertase molecule at the site, making C5 convertase. C5 convertase has specific enzymatic activity to cleave C5 (190 kD), releasing both the smaller C5a (11 kD) fragment (a 74 amino acid glycopolypeptide) and the C5b (180 kD) fragment into solution.* The subsequent assembly of the MAC is nonenzymatic. Fluid-phase C5b binds first C6 (120 kD) and then C7 (110 kD), forming a stable C5b67 complex. The binding of C7 converts the complex from a hydrophilic to a hydrophobic state, which then preferentially inserts the complex into lipid bilayer – including other cell membranes in the immediate vicinity of the primary surface on which complement activation is focused. C8 (152 kD) then binds to the C5b67 complex at a site on C5b, forming C5b678 as it inserts itself into the lipid bilayer membrane. Finally, the C5b678 complex induces C9 polymerization [1627] into the form of a hollow tubular structure, with 12-18 C9 monomers (69 kD) attached to each C5b678 complex [1613, 1629], completing the MAC. Poly(C9) is a cylinder with inner and outer diameters of 9 nm and 15 nm respectively, tube length 15 nm, rimmed by a 4.6 nm thick torus with inner and outer diameters of 11 nm and 22 nm on one end [1628, 1629].
* C5b also appears to enhance the phagocytosis of yeast [1620].
The MAC [1613-1616], a dimer of the ~1650 kD [1630] C5b6789 complex, makes a single transmembrane channel through which water and electrolytes may pass [1631], resulting in an impairment of osmotic regulation and subsequent cytolysis [1632]. This is similar to the action of mammalian cytolytic T lymphocytes that can kill targeted cells by inserting into their membranes a 67-kD pore-forming molecule called perforin [1633-1635] which has structural homology to C9 [1634, 1638]. Similar molecules are found in the granules of eosinophils [1639], various bacterial pathogens [1635-1637], and in the protozoan parasite Trypanosoma cruzi [1640]. Complement-mediated lysis has been shown for many kinds of cells including erythrocytes, platelets, lymphocytes, bacteria, and viruses possessing a lipoprotein envelope [1617]. S. Flitman notes that flares of autoimmune diseases like lupus produce clinically detectable drops in C3 and C4 levels.
Despite being targeted principally against microbial intruders, complement is relevant to nanorobotic nanomedicine because several possible nanorobot building materials are already known to interact with components of the complement system. For example, graphite adsorbs C1q and C3 (Section 15.3.3.3) and Teflon activates C5a (Section 15.3.4.3). Alumina ceramic (sapphire) has not yet been found to activate complement [613] or complement receptors [1641], but some fullerenes can induce the production of specific IgG antibodies [724, 725] which could enable complement activation along either pathway. Diamond may adsorb some C3 like many other hydrophobic surfaces (Section 15.3.1.1), though diamond is generally considered noninflammatory relative to the complement system. In one experiment [1642], diamond particles caused insignificant complement activation, unlike crystals of monosodium urate monohydrate, hydroxyapatite, brushite, and calcium pyrophosphate dihydrate, and particles of blackthorn, all of which demonstrated activation of C3 via the alternative pathway as determined by immunofixation following electrophoretic separation of C3 and its activation products.* Complement activation at the site of surgical trauma has also been reported during cardiac surgery [4953-4960].
* Urate crystals directly activate [2327, 2328] and amplify [2322] the classical complement pathway, induce alternate pathway activation when the classical pathway is inhibited [1648], and promote C5a production via assembly of a stable C5a convertase on the crystal surface [2522].
There are three principal physiological consequences of complement activation [1613], all of which are directly relevant to nanomedicine. Nanorobots could potentially activate complement causing (1) inappropriate cell death, (2) release of vasoactive substances and shock, or (3) stimulation of an autoimmune-type response, as follows:
First, the cytolytic MAC is assembled on the target surface. This may not be a direct threat to hard nanorobot targets, whose exteriors typically will be dissimilar to those of foreign cells or viruses and may be made of tough materials impossible for the MAC to breach. However, the MAC can attack nearby native cells as well as foreign cells (e.g., in cell transplants), causing undesirable cellular necrosis [234]. Native blood cells have some protection against such attack. For instance, an average of 25,000 MACs can be assembled on a neutrophil surface without lysis because MACs are rapidly shed with a clearance half-life of ~2 minutes at 37 oC [1692] – about two-thirds of the MACs are ejected from the cell in plasma membrane vesicles via an exocytic process, and one-third are removed via endocytic internalization and proteolysis. Similar processes are observed on platelets [1693] and other native cell types [1694, 1695].
Second, the many short peptide cleavage fragments resulting from the sequential complement activation chain may induce potentially harmful side effects. These effects may include: (1) local inflammation (Section 15.2.4) (e.g., via C2b, or C2 kinin after plasmin modification), (2) inhibition of the growth of antigen-antibody complexes (e.g., via C3a) [234, 5928], and (3) anaphylactic reaction (Section 15.2.6.1) to C3a and C5a (and to a lesser extent C4a) which are themselves potent anaphylatoxins that bind to mast cells, degranulate mast cells and basophils, and induce release of vasoactive substances like histamine that mediate vasodilation, increased vascular permeability, and contraction of bronchial smooth muscle.
Third, complement cleavage fragments also include chemotactic factors such as C3a, C5a, and soluble C5b67 complex. These factors attract neutrophils, macrophages, and other phagocytic cells to the vicinity, thus increasing the opportunity for phagocytic uptake of nanorobots by these protective native cells. Macrophages display receptors* for C3b on their surfaces, enhancing uptake of non-self particles bound to C3b. Soluble C5b67 released into solution phase as a result of either the presence or the activities of medical nanorobots could enter the membranes of nearby uninvolved cells, possibly leading, eventually, to cytolysis of those nearby cells.
* Four commonly known receptors for C3b and C4b are CR1, CR2, CR3, and CR4, some or all of which are found on B lymphocytes, epithelial cells of cervix and nasopharynx, erythrocytes, follicular dendritic cells, glomerular epithelial cells, macrophages, monocytes, neutrophils, and NK cells [1618]. Bacteria opsonized with antibody and complement are often observed adhering to human red cells [1618].
The ideal medical nanorobot design would include an exterior surface that does not activate complement [5825]. It is possible that a pure diamond or sapphire surface will not activate complement. However, nanorobot exteriors may need to display sorboregulatory or adhesioregulatory (Section 15.2.2.4), anti-inflammatory (Section 15.2.4), or antithrombotic (Section 15.2.5) ligands whose complement activation potentials have not yet been widely studied. One approach might be for nanorobots to mimic autologous human self-surfaces [1613, 1625], that contain molecules of CR1 (a natural C3b receptor) and/or membrane cofactor protein (MCP) [1685] that bind to C3b and also promote the preferential binding of factor H (see below) rather than factor B to C3b [1618]. This would effectively limit C3 deposition and prevent the formation of stable C3 convertase enzymes. By comparison, non-self-surfaces allow the rapid deposition of many molecules of C3 [1618]. The additional presence of decay accelerating factor (DAF) [1643, 1684] on self-surfaces, along with CR1, is known to inhibit the association between C3b and B, and to promote dissociation of the C3bBb complex [1618]. Membrane sialic acid also appears to be one of the carbohydrate components protecting autologous cell membranes from amplified C3b deposition [1618]. Malignant tumor cells are observed to use membrane-bound complement regulatory proteins to evade complement-mediated injury [1644].
Other complement-resistant surfaces have been investigated. For instance, polymers containing phosphorylcholine polar groups can achieve a marked reduction in complement activation as measured using radioimmunoassay for C3a [578]. Another investigation [1645] of biomaterial-mediated complement activation used an animal implantation model and gold surfaces bearing various thiol-linked functionalities. This study found that mercaptoglycerol- and mercaptoethanol-bearing surfaces engendered the strongest inflammatory responses (as reflected by the accumulation of large numbers of adherent neutrophils and monocytes/macrophages) whereas L-cysteine-coated surfaces caused only minor inflammatory responses. Both glutathione-modified and untreated gold implants attracted minimal numbers of inflammatory cells [1645]. The mercaptoglycerol surface – which has hydroxyl groups (alternative pathway) and high IgG affinity (classical pathway) – caused pronounced production of C3b and C5b6789 in serum and increased C3 deposition on the surface. By comparison, bare gold surface and mercaptopropionic acid surface caused very little complement activation [1646]. Particle surface coatings of PLA-PEO diblock copolymer exceeding ~0.20 molecules/nm2 significantly reduce complement opsonization of PLA-PEO nanoparticles [2487]. Acceptable levels of complement activity reduction may be determined experimentally. Generally, surfaces with negative or neutral charge do not activate complement, as compared to positively charged surfaces.
If complement-active surfaces cannot be avoided, active nanorobots may interrupt the complement activation process in their vicinity via controlled emissions of one or more of the >11 activation control proteins [1613, 1622, 1647], by depleting essential factors, or by other means, as may be appropriate for the particular mission and nanorobot design. Complement is continuously activated in the body, both in health and in disease – endothelia, circulating cells, and other plasma-exposed tissues are under constant attack [1613]. To prevent significant damage to self-cells, the complement system must be tightly regulated.
On the classical pathway, human C1 esterase inhibitor (C1 INH) [1620] or C1 inactivator (C1 INA) [1648] is a naturally-occurring heat- and acid-labile serum alpha2-neuraminoglycoprotein (MW = 109,000 daltons) that stoichiometrically binds C1 and inhibits not only C1r and C1s, but also plasma kallikrein, plasmin, trypsin, chymotrypsin, activated Hageman factor (clotting factor XIIa), and activated thromboplastin antecedent (factor XIa) [1617]. Normal plasma concentrations of C1 are ~6 x 10-5 gm/cm3 (~360 molecules/micron3) in human blood (Appendix B). A release rate of ~103 molecules/sec (~0.0002 micron3/sec) would maintain an equal concentration of C1 INH in a 1-nm skin layer around a 1-micron spherical nanorobot washed by a 1 mm/sec capillary blood flow. Natural serum also contains a glycoprotein called C4bp or C4b-bp (540 kD) [1649] that has a specific binding affinity for C4b (e.g., it competes with C2a for binding to C4b) and is the control protein for the classical C3 convertase.
On the alternative pathway, factor H (beta1H or C3b INA accelerator, 150 kD) [1650] can stabilize C3b and prevent its interaction with factor B. The C3b-factor H complex is then cleaved by factor I (C3b/C4b inactivator or C3 INA, 93 kD), for which MCP (membrane cofactor protein) is a necessary cofactor [1620, 1685]. DAF (see above) is a surface-bound glycoprotein that accelerates the decay of both classical and alternative pathway C3 convertases [1620, 1643, 1684]. Nanorobots could also employ molecular sorting rotors (Section 3.4.2) to deplete [1677-1679] local supplies of activated factor D (MW = 22,000 daltons, serum conc. ~ 1.5 x 10-6 gm/cm3 or ~40 molecules/micron3 [1617, 1677]), or to deactivate local factor D molecules using a catch-and-release process. (While it might appear harmful to inactivate complement in a septic patient, this inactivation is temporary and would occur only as a part of a comprehensive nanorobotic-based anti-infective treatment.) A similar process is employed by neutrophils to deactivate anaphylatoxins. For instance, neutrophil surfaces include 50,000-113,000 receptors (C5aR, MW ~ 40-60 kD) for C5a [1618]. Following receptor binding, the C5a is internalized and degraded to inactive peptide fragments [1618]. A related strategy is to release solubilized complement receptors to deplete complement components [1655].
Farther downstream, plasma enzyme carboxypeptidase N (CPN) or anaphylatoxin inactivator (AI) (280 kD) abolishes the activities of C3a and C5a by removing the C-terminal arginine from both molecules [1617]. The 56-kD serratial protease eliminates C5a chemotactic activity at a dose of 1 µg/cm3 (~10 molecules/micron3) [1651]. Still farther downstream, S-protein (vitronectin, 83 kD) binds to fluid-phase C5b67, preventing its insertion into lipid bilayers [1618]. The regulatory glycoprotein clusterin (~80 kD) serves a similar function [1686-1689] and is a more active inhibitor on a molar basis, but the effects of the two inhibitors are additive [1686]. Autologous cells also have two MAC-inhibiting proteins, called homologous restriction factor (HRF C8-binding protein, 65 kD) [1652] and 20-kD glycoprotein CD59 [1653], which protect them against lysis by the MAC [1618]. CD59 is present on all circulating cells, endothelia, epithelia, and in most organs [1613] – erythrocytes display ~25,000 copies/cell [1690] and many nucleated cell types express much more. But interrupting the cascade this far downstream cannot prevent the adverse inflammatory and chemotactic effects of the upstream peptide cleavage fragments.
Control protein-oriented strategies have already been pursued experimentally. In one study [1654], the modification of reactive surface hydroxyl groups on regenerated cellulose with various dicarboxylic-acid anhydrides was found to significantly limit the complement-activating potential of these materials. Maleic anhydride displayed the most dramatic and consistent diminution of complement activation compared to unmodified cellulose (i.e., 0-10% of control values for C3b deposition and C3a/C5a production [1654]). This maleated derivative was found to facilitate the factor H control of C3 convertase and C5 convertase activity, thus limiting complement activation and the production of other inflammatory mediators via the normal regulatory mechanisms. A chimeric molecule combining DAF and CD59 retained the inhibitory activities of both component molecules [1709].
Monoclonal antibodies have been raised against C4 [1710], C5 [1711, 1712], C5a [1713, 1714] and C5a receptor [1715], C6 [1716], and C8 [1711], and peptide antagonists to C5a receptor have been tested in vitro [1717-1721]. A variety of therapeutic complement inhibitors are under active investigation [1655-1657, 5348], including an RNA aptamer [5492] inhibitor of C5 [1722]. Heparin also inhibits complement activation [2485] but would not be a particularly viable option here due to its principal activity as a potent anticoagulant.
As might be expected, bacteria have evolved many techniques of evading complement activation. Some of these, in principle, could be mimicked by medical nanorobots. (Bacteria that are not killed and lysed in serum by the complement MAC are said to be serum resistant. Many of the Gram-negative bacteria that cause systemic infections (e.g., septicemia) are serum resistant.) For example:
(1) Brucella abortus bacteria may use O-antigen to shield outer membrane proteins from C1q binding [1671];
(2) the bacterial capsule of Neisseria meningitidis contains sialic acid (a common component of host cell glycoproteins) which inhibits C3b opsonization and inhibits activation of the alternative pathway [1658-1661];
(3) Helicobacter pylori urease is believed to degrade bound C3b, reducing opsonization [1672];
(4) YadA protein produced by Yersinia enterocolitica binds factor H, reducing C3b deposition on the bacterial surface probably by rapid inactivation of C3b [1668];
(5) Neisseria meningitidis and Haemophilus influenzae (which cause bacterial meningitis) can covalently attach sialic acid residues to the O-specific sugar portion of LPS, preventing the formation of C3 convertase and thus imparting resistance to MAC [1437];
(6) lipooligosaccharide sialylation of serum-sensitive N. gonococci in vivo converts them to serum-resistant [1659, 1662];
(7) Pseudomonas aeruginosa produces extracellular elastase and alkaline protease enzymes that inactivate components of complement [1663-1666];
(8) a protease produced by Bacteroides gingivalis inactivates C3 [1667];
(9) streptococcal M protein binds factor H, inhibiting complement activation [1669, 2517];
(10) group A and B streptococci express on their membranes an enzyme that cleaves 6 amino acids from C5a, rendering this agent inactive [1691];
(11) 17-kD outer-membrane protein Rck promotes resistance to complement killing of Salmonella typhimurium by interfering with C9 polymerization [1670];
(12) some bacteria with LPS molecules having long intact O-antigen side-chains can prevent effective complement killing by holding the MAC complex too far from the vulnerable outer membrane to be effective [1673, 1674].
Numerous viruses have acquired host complement regulators, especially CD59 [1696-1702], glycoprotein C (gC) [2344] which is found in the envelopes of herpesviruses HSV-1 and HSV-2 that binds to C3b, and glycoprotein III (gIII) [2345] which is found on pseudorabies virus and serves a similar function. Several parasites have been reported which express surface molecules that inhibit MAC formation, particularly Schistosoma mansoni [1703-1705], Trypanosoma cruzi [1706], and Entamoeba histolytica [1707, 1708].
Last updated on 30 April 2004
{kind=link}
{kind=link}