Nanomedicine, Volume IIA: Biocompatibility
© 2003 Robert A. Freitas Jr. All Rights Reserved.
Robert A. Freitas Jr., Nanomedicine, Volume IIA: Biocompatibility, Landes Bioscience, Georgetown, TX, 2003
15.2.5 Coagulation and Thrombogenicity
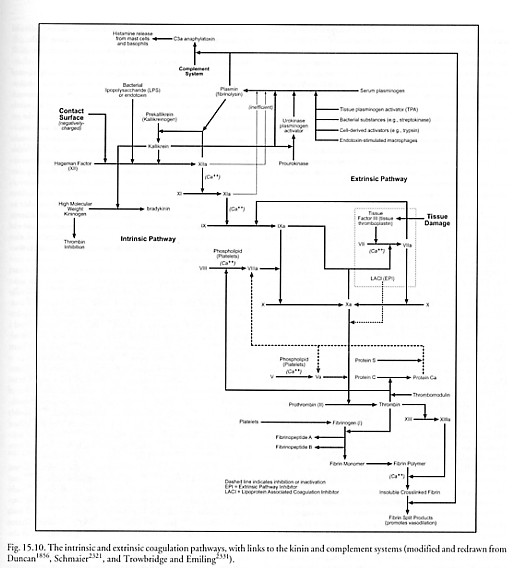
Blood coagulation involves a complex series of reactions in which various proteins are enzymatically activated in a sequential manner, transforming liquid blood into a gel-like mass which is then stabilized to form a thrombus (clot) consisting of platelets, fibrin, and red cells. Mechanical blockage by fibrinogen clots helps prevent the spread of microbial invaders. The series of reactions (Figure 15.10) is classically divided into two pathways – extrinsic and intrinsic – involving more than a dozen factors, that converge on a single common final pathway resulting in clot formation [234, 1753, 1856, 1859-1863].
The extrinsic pathway is initiated at the site of tissue injury with the release of tissue factor (factor III or tissue thromboplastin) [1864] which is found on the surfaces of many extravascular cells. In the presence of Ca++ ion, factor III activates factor VII to VIIa which then activates factor X [1865-1867] to Xa, initiating the common final pathway.
The intrinsic pathway is initiated by activation of the contact factors by a negatively charged surface [1840-1845] – e.g., tissue material such as collagen on the exposed surface of a blood vessel, in vivo [234], or glass [1839] or particulate material such as kaolin [1857] or urate crystals [2327, 2329], in vitro. The intrinsic pathway begins with human factor XII (FXII or Hageman factor), a serine protease produced by the liver that circulates in plasma as an 80-kD single-chain inactive zymogen. The zymogen is activated via (1) interaction with negatively charged surfaces, (2) bacterial LPS [2332], or (3) via proteolytic cleavage by kallikrein (produced from prekallikrein by plasma kininogen [1860, 2320, 2321]). After this activation, the zymogen is proteolyzed by the kallikrein into a two-chain active protease, activated FXII (FXIIa) [1868]. FXIIa can activate several plasma cascade systems [1853] including the contact system [1843, 1869-1871], fibrinolysis, and the complement system (Section 15.2.3.2) as well as the intrinsic pathway. FXIIa attacks prekallikrein to generate more kallikrein, setting up a positive feedback (amplification) control loop. FXIIa also activates contact factor XI to XIa in the presence of Ca++, and the additional kallikrein releases bradykinin (a polypeptide with potent vasodilator and pain-producing action) from kininogen. Factor XIa, again in the presence either of Ca++ or of factor VIIa from the extrinsic system, activates factor IX to yield the serine protease factor IXa. Factor IXa then cleaves a bond in factor X [1865-1867] to produce the 2-chain serine protease, factor Xa, in the tenase complex (VIIIa, IXa, X and Ca++) on the surface of activated platelets, initiating the common final pathway.
In the common final pathway, factor V [1872-1874] is activated to factor Va in the presence of Ca++ by trace amounts of thrombin [1875-1878]. Factor Va then interacts with Xa and platelet anionic phospholipids on the surface of activated platelets to convert prothrombin to thrombin, a serine protease of the trypsin family. This more abundant thrombin produces more Va from V and converts VIII to VIIIa, XI to XIa, and XIII to XIIIa. The last step in the sequence is the proteolytic cleavage of fibrinogen by thrombin (a 34 kD, ~4.6 nm diameter roughly spherical molecule [1879]), leading to the release of two fibrinopeptides, A and B, and the production of fibrin monomer. The fibrin monomers are polymerized and crosslinked by activated factor XIIIa in the presence of Ca++, producing a stable insoluble polymer and a clot. After the clot has formed, it can later be dissolved during fibrinolysis: inactive plasminogen (90 kD) is cleavage-activated by tissue plasminogen activator (tPA) or urokinase to release a serine protease, plasmin, which can cleave the fibrin polymer.
For coagulation to occur, platelets must undergo adhesion and activation [6043]. The adhesion of platelets to exposed collagen in injured blood vessels is mediated by a bridging molecule called von Willebrand’s factor [1880] that is secreted by endothelial cells into plasma. This prevents platelets from detaching under the high shearing stresses developed near vessel walls. The activation of normally quiescent platelets is a complex phenomenon that includes changes in cell shape, increased movement, aggregation, and release of the contents of their granules containing nucleotidyl phosphates, serotonin [1881], various factors, enzymes and plasma proteins. The most potent activator of platelets in vivo is thrombin [1882]. Thrombin interacts with a receptor on the platelet plasma membrane, followed by transmembrane signaling and subsequent activation of the cell. Collagen [1883] is the other most important platelet activator. ADP can stimulate aggregation but not granule release.
In principle, the blood-contacting surfaces of a nanoorgan, or of nanorobots [22] in sufficient bloodstream numbers and concentrations, could activate platelets or either of the two coagulation pathways. That is, a poorly-chosen nanodevice exterior exhibiting negatively charged surfaces (Section 15.5.6.2) could contact-activate the intrinsic pathway, or careless mechanical actions by in vivo nanodevices could cause tissue injury to extravascular cells sufficient to invoke the extrinsic pathway. Careful choices of materials and of allowable mechanical motions (Chapter 15.5) should reduce or eliminate inherent nanodevice thrombogenicity and red cell hemolysis (Section 15.5.5.1.1). The fact that natural endothelium is nonthrombogenic [5961] provides an existence proof that such surfaces can exist, and strongly suggests that it should be possible to bioengineer [5962] or nanoengineer such surfaces from artificial materials, including active components providing metered emissions of useful antithrombogenic mediators [5963]; marrying natural endothelium to artificial surfaces [5964], or “endothelialization,” is well-known in vascular grafting [5965-5971].
For example, DLC diamond-coated stents [626, 628, 4723], heart valves [612] and other blood-contacting LVAD (left ventricular assist device) surfaces [596, 613, 1680] and substrates [597, 660, 4726, 4730] generally show reduced thrombogenicity and weak or no platelet activation [660, 4726]. Pyrolytic carbon (LTIC) may be somewhat thrombogenic during brief exposures to blood [814]. But LTIC is considered a fairly nonthrombogenic material (with relatively low platelet adherence [1680]) for long-term exposures to blood [808] such as in heart valves [813], especially if very pure [908]. Fullerene thrombogenicity is unknown, though several forms of graphite are somewhat thrombogenic [819, 822]. Carbon composites show at least short-term thromboresistance [829], though with some surface accumulation of platelets [830]. Carbon black particles can produce prompt thrombocytopenia [875, 884] along with cerebral thromboemboli [884], possibly due to uncontrolled surface chemisorption effects. Platelets adhere less readily to Teflon after longer exposures [1202, 1326] but their reactivity may be enhanced [1159], which suggests that bulk Teflon is thrombogenic [1192, 1195, 1317, 1326]. There is also contrary evidence [1192, 1209], possibly due to the many different forms of the material in use. Albumin-bound Teflon [1328, 1330] and low-roughness surfaces [1315] may be moderately thromboresistant, but Teflon prostheses [1370], catheters [1375], and tubes [1189] have produced significant thromboses. Sapphire (alumina ceramic) has low thrombogenicity [1058-1060] and both platelet adhesion [977] and activation [1060] are low. Hemolysis (Section 15.5.5.1.1) is near-zero for diamond [643, 660, 4726], graphite [643], and alumina [643] powders, though Teflon patches used to repair atrial septum defects in the 1970s were sometimes mechanically hemolytic [1347, 1348].
Future experiments must determine if ordinary diamondoid surfaces will have to be supplemented with additional antithrombogenic coatings in order to achieve medical nanorobot mission objectives. If such coatings are required, one simple possibility is surface-immobilized heparin, a ~15 kD straight-chain anionic (acidic) mucopolysaccharide (glycosaminoglycan) that forms polymers of various lengths. Heparin, first discovered in 1916 [2364], is produced naturally by human liver mast cells and basophil leukocytes. It inhibits coagulation primarily by enhancing about ~1000 times the ability of antithrombin to inactivate a number of coagulation enzymes, including thrombin and activated factors X, XII, XI, and IX [5489]. Nanorobot exteriors could be “heparinized” [1884-1891] and thereby rendered thromboresistant by immobilized heparin on all blood-contacting surfaces at ~monolayer surface concentrations (e.g., 7-10 pmol/cm2 [1891]). Cellulose membranes coated with 3.6 pmol/cm2 of endothelial-cell-surface heparin sulfate show complete inhibition of platelet adhesion [1892]. Albumin-heparin conjugate coated surfaces also display anticoagulant activity [1893] – pre-adhered endothelial cells proliferating on this coating significantly reduce the number of platelets which subsequently adhere to the surface, and other immobilized heparin conjugates have also given promising results [1894]. Unfortunately, heparin can have undesirable side effects such as binding with platelet factor 4, which then induces associated antibody production leading to thrombocytopenia [2366, 2367], so a synthetic heparin-like analog may need to be engineered with properties similar to low molecular weight heparin (e.g., enoxiparin [5495]) which decreases risk [5489] of HITT (heparin-induced thrombosis) syndrome [5496]. Coatings of hydrophilic acrylic copolymers with salicylic acid residues have also given good antithrombogenic behavior in animal studies [1895], and hirudin-thrombin complex adsorbed on glass bead surface did not stimulate fibrinogen activation [1896]. Polyethylene electret film has even been studied for its athrombogenic properties [1897].
If satisfactory passive nonthrombogenic surfaces cannot be found, nanorobots might employ any of at least four active strategies to prevent iatrogenic coagulation:
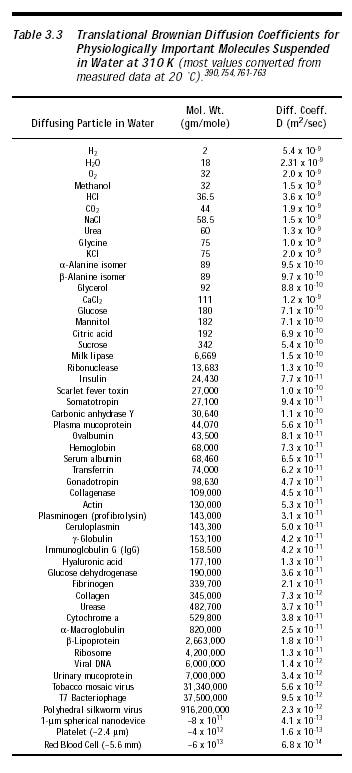
(1) Local Factor Depletion. Factor X (56 kD) and factor V (330 kD) are the only two coagulation factors whose removal would interrupt both intrinsic and extrinsic pathways prior to the deposition of fibrin. Conveniently, these two factors have among the lowest blood concentrations of any of the factors – 0.005 mg/cm3 (~50 molecules/micron3) and 0.005-0.012 mg/cm3 (9-20 molecules/micron3), respectively (Appendix B), though factor X is the preferred target because factor V is also present in platelets. The diffusion limit of 56 kD molecules having diffusion coefficient ~8 x 10-11 m2/sec (Table 3.3), if present at a concentration of ~10 molecules/micron3, is ~104 molecules/sec delivered to the surface of a spherical nanorobot 1 micron in radius (Section 3.2.2). Nanorobot surface-deployed sorting rotors (Section 3.4.2) could selectively deplete factors X and V from serum at the rate of ~10 molecules/sec-rotor (Section 4.2.3), eliminating coagulability in a hematologically isolated local environment or greatly reducing coagulability if fresh blood is continuously replenishing the factor supply. Interestingly, an experiment using gold-coated polyurethane film chemisorbed with any of three different peptides found that the film acted as a thrombin scavenger, absorbing thrombin with high affinity and selectively removing it from plasma [1898], a crude analog of the more sophisticated procedure described above.
(2) Global Factor Depletion. Comprehensive management of the entire bloodstream inventory of coagulation factor molecules with whole-body real-time control also appears feasible nanorobotically, unlike contemporary heparin or warfarin anti-coagulation agents. There are ~1017 molecules each of factor X and factor V present in free form in the whole blood volume.* A population of 1012 bloodstream-resident 10-micron3 nanorobots each having 104 sorting rotors on its exterior surface would require ~10 sec to remove ~90% of the entire serum inventory of either factor. (The removal rate is restricted by the maximum diffusion limit.) Each molecule would receive some minor chemical modification that inactivates it before it is released back into solution, to preclude the need for onboard storage. Prothrombin (72 kD, ~800 molecules/micron3 in serum; Appendix B) could also be selectively depleted from serum, either locally or globally, before it can be cleaved to make activated thrombin. Even thrombin itself (34 kD, ~0.3 molecules/micron3 basal [1899] to ~700-900 molecules/micron3 thrombotic [1900]) could be nanorobotically depleted, chemically modified, and then released. Note that this method is unsuitable if the patient is bleeding and requires prompt hemostasis, except possibly for therapeutic nanorobots deployed in conjunction with clottocytes [22] (Chapter 24). Factor depletion seems most appropriate as a temporary measure to avoid nanorobot-induced thrombogenesis in hematologically intact patients.
* The presence of Ca++ ion is a crucial ingredient in at least six enzymatic steps of the coagulation cascade. Reducing Ca++ to minimal levels near the nanorobot would effectively prevent coagulation in the local vicinity, or greatly reduce it if there is exogenous replenishment. The normal concentration of serum or extracellular Ca++ is quite high compared to coagulation factors, ~106 ions/micron3 (Section 10.4.2.1). Still, 104 surface-resident Ca++ sorting rotors per nanorobot could remove ~1010 ions/sec (Section 3.4.2) from the local environment, thus depleting the nearest 10,000 micron3 of plasma of all Ca++ ions. Those 1010 ions could then be stored in ~5% of the internal volume of a 10-micron3 nanorobot. A bloodstream population of ~0.5 trillion of such nanorobots could reduce serum Ca++ concentration to <1% of normal in ~1 sec. If chelated (e.g., citrated) and released, the Ca++ would be temporarily unavailable to the coagulation cascade because the ion is tightly bound, although citrate is rapidly metabolized by the body, freeing the Ca++. J. Rootenberg cautions that mission design should include analysis of whether these local actions might induce trans-cellular stasis reactions. R. Bradbury notes that chelating serum Ca++ would likely disrupt many biological processes, and might even induce release of mitochondrial Ca++ stores; use of EGTA might provide longer-term chemical sequestration of Ca++.
(3) Inhibitor Release. Instead of depleting coagulation factors, nanorobots could release coagulation inhibitors [2318, 2319] during the nanomedical mission, either locally or globally, and then retrieve these molecules before exiting the body. The simplest approach is to inhibit thrombin, the cornerstone molecule of the coagulation cascade. There are four naturally occurring thrombin inhibitors found in normal plasma [1753] – antithrombin III (potentiated by acidic proteoglycans such as heparin), alpha2-macroglobulin, heparin cofactor II, and alpha1-antitrypsin (alpha1-antiproteinase). Various other thrombin inhibitors are also known [1907-1910, 1914-1920] including most especially the hirudins [1909-1913]. There are a number of factor X inhibitors, including the coumarin drugs (e.g., warfarin [1935-1937]), low-molecular-weight (4-6.5 kD) heparins [1901-1903] such as a heparin pentasaccharide with purely anti-factor Xa activity [1904, 1905], and vast numbers of other alternatives [1921-1935, 2318, 2319] including heparin mimetics [2365] that avoid heparin’s unwanted side effects. For example, synthetic factor Xa inhibitor FX-2212 inhibits Xa activity by 50% at a serum concentration of 272 nM (164 molecules/micron3) in vitro [1927]. Both indirect [1938, 1939] and direct [1940-1949] inhibitors of factor V or Va have been reported [1950], and prothrombin activation inhibitors are known [1951-1957]. Inhibitory monoclonal antibodies [3962] have also been raised against several of the coagulation proteins.
Nanorobots could also release any of a number of platelet inhibitors to prevent coagulation. Platelet adhesion inhibitors are well known [1958-1961, 1968]. Persantine [382], prostacyclin [1962], ibuprofen [1962], and even nitric oxide [1963] have a demonstrable effect on platelet deposition. Platelets can be prevented from adhering using an RGD (Arg-Gly-Asp) tripeptide-containing peptide that acts as an antagonist for the fibrinogen receptor on platelet surfaces [1964, 1965], e.g., when administered at ~0.6 ng/sec per cm3 of blood in live dogs [1964]. Platelet activation inhibitors are also well known [1966-1969] and include nitric oxide [1970], prostacyclin [1971], kininogens [2321], and artificial peptides [1972]. Platelet degranulation inhibitors have been investigated [1973-1978]. Platelet aggregation inhibitors include kininogens [2321] and a wide range of anti-aggregating drugs [1968, 1979-1985] such as aspirin [1986], clopidogrel [1987], ticlopidine [1988, 1989], crotalin (inhibitory dose ~10-6 gm/cm3 or ~100 molecules/micron3 in mouse serum [1990]), adamantane derivatives [5572], and potent natural aggregation inhibitors such as prostacyclin [1991]. One potential difficulty with this approach is that most of the enzyme cascade reactions take place in complexes on surfaces, and the spatial arrangement of clotting factors [1992] may prevent the inactivation of factors by nanorobot-released inhibitors, proteolytic enzymes, or specific antibodies unless those molecules are applied locally. (A. Kumar notes that global neutralization of clotting factors could increase the risk of petechiae and microbleeds for the duration of the nanorobotic mission, possibly producing further complications for the patient such as loss of blood volume or edema.)
(4) Regulatory Control. The coagulation system is a complicated cascade of enzymatic reactions. Feedback mechanisms provide a delicate balance of activation and inhibition at each point in the cascade. Fibrin clots are constantly being laid down and dissolved in a state of dynamic equilibrium. Medical nanorobots may cautiously intervene in, and possibly manipulate, this dynamic control process. For example, protein C and protein S are two vitamin K-dependent coagulation proteins that provide a vital control mechanism in the cascade. Protein C is activated to Ca (activated protein C or APC) by thrombin (with thrombomodulin). But then this activated protein Ca (with cofactor protein S) inactivates the activated factors Va and VIIIa by proteolytic degradation [1993, 1994], which in turn inhibits the formation of thrombin via factor Xa. Protein S is itself cleaved and inactivated by factor Xa [1995] and has Ca-independent anticoagulant activity [1999], and of course there is also a protein C inhibitor [2000]. A deficiency of either C or S is associated with venous thromboembolism. It is possible that an artificial surplus of protein Ca [2001, 2002] and protein S could significantly brake the coagulation process. (Natural protein Ca circulates at 3-5 mg/liter [1996] or ~30 molecules/micron3 in human blood with a half-life of ~1000 sec [1997].) Similarly, a factor XIIa inhibitor [1998] might inhibit the intrinsic pathway from the top; tissue pathway factor inhibitor (TPFI) might interrupt the extrinsic pathway in some instances [1864, 1922]; and so forth. The medical nanorobot designer should verify that no chemical substance displayed or emitted by the nanorobot will mimic the structure or activity of natural thrombotic stimulators or key coagulation factors such as tissue thromboplastin, factor Xa, or thrombin.
The possible risk of nanorobot-induced bleeding is discussed in Section 15.6.2.
Last updated on 30 April 2004
{kind=link}
{kind=link}